What is Beta-Thalassemia?
Beta-thalassemia is an inherited blood disorder characterized by low levels of hemoglobin, leading to anemia and insufficient oxygen supply to the body. Common symptoms include jaundice, fatigue, organ damage, and delayed growth and development.
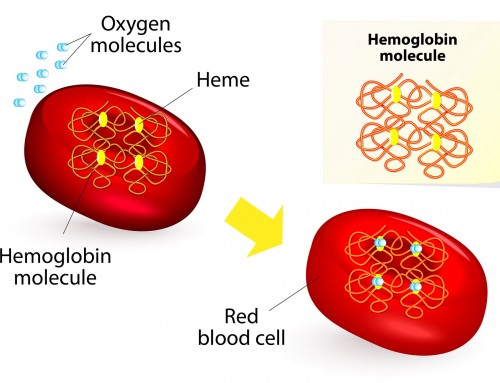
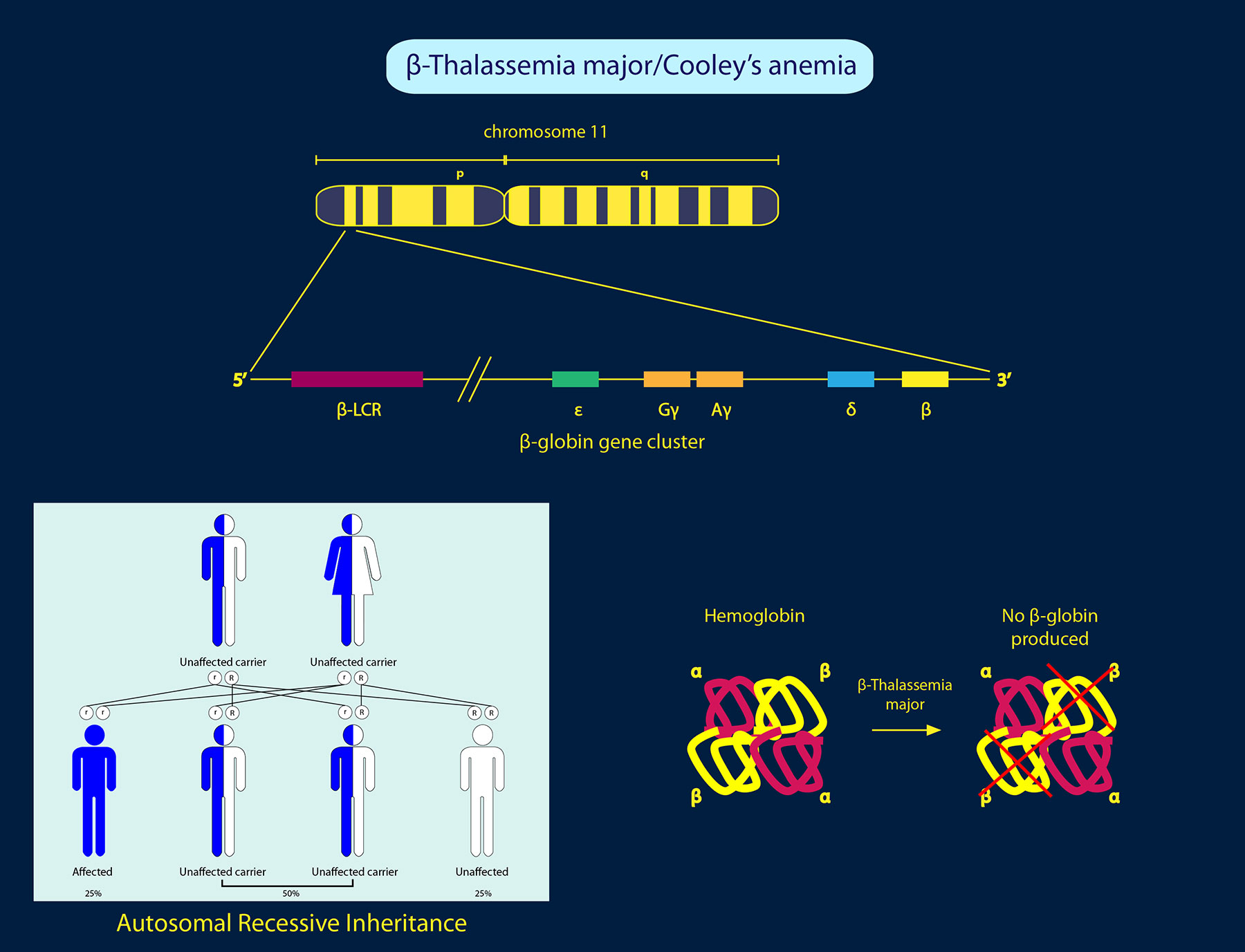
Hemoglobin is an iron-containing protein in red blood cells that carries oxygen to the rest of the body. Normal hemoglobin (HbA) is made up of four protein subunits: two α-globin chains and two β-globin chains. Beta-thalassemia is caused by genetic mutations in the HBB gene, which encodes the hemoglobin β-globin subunit.
Beta-thalassemia is classified into 3 types based on the clinical severity of the symptoms. The three types of beta-thalassemia are:
1. β-thalassemia major –Two copies of the HBB gene are defective in this type beta-thalassemia, also known as Cooley’s anemia. Symptoms such as severe anemia, hypochromic (paler than normal) and microcytic red blood cells are observed, and they can become life threatening if left untreated.
2. β-thalassemia intermedia – Two copies of the HBB genes are affected, but the symptoms can vary from mild to severe depending on the amount of β-globin produced.
3. β-thalassemia minor – Only one copy of the HBB gene is affected, and individuals are known as carriers of beta-thalassemia. They usually do not show symptoms, but some may have mild anemia.
References:
Origa R (2000, Updated 2015 May 14). Beta-Thalassemia. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2016.
Cao A, Galanello R (2010). Beta-thalassemia. Genetics in Medicine 12, 61–76;
Muncie HL, Campbell JS (2009). Alpha and Beta Thalassemia. Am Fam Physician. 80(4): 339-344.
DNA In the News2017-04-06T20:19:29+00:00