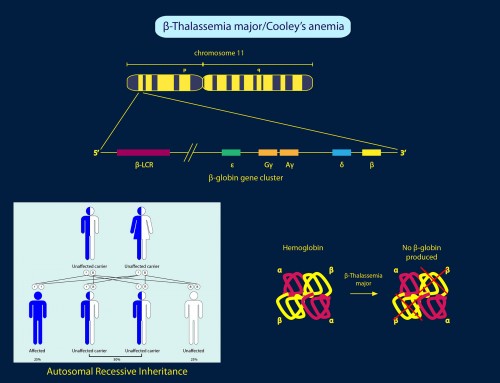
The signs and symptoms of beta-thalassemia can vary in severity depending on the extent of
HBB gene deficiencies. The severity of the disease is classified into three types in the order of decreasing severity: β-thalassemia Major, β-thalassemia Intermedia and β-thalassemia Minor.
β-thalassemia Major
People with β-thalassemia Major usually have no symptoms at birth, but signs and symptoms often appear between 6-24 months of age. This is due to the presence of fetal hemoglobin (HbF) remaining at birth, which can mask the deficiency until the body switches to adult hemoglobin (HbA) synthesis.
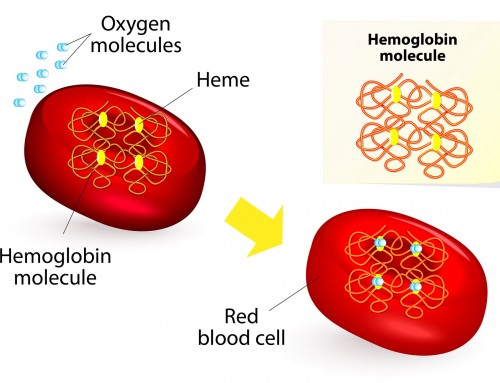
β-thalassemia Major is the most severe form of the disease. Affected people develop life-threatening anemia, a condition where the amount of red blood cells circulating in blood is lower than normal. As a result, insufficient oxygen and nutrients are supplied to the body, resulting in fatigue, growth retardation and often delayed puberty. They may also develop dark urine, jaundice, misshapen bones and progressive enlargement of the abdomen caused by the spleen and liver. Regular blood transfusions are often required in order to replenish their red blood cell supply.
Complications that can arise from β-thalassemia Major include:
- Excess iron (usually caused by repeated blood transfusions, resulting in iron overload and damage to organs and tissues)
- Bone deformities and broken bones (caused by expanded bone marrow, resulting in bones that are more brittle, leading to breakage or abnormal bone deformities)
- Enlarged spleen (caused by the spleen working harder to filter out dead red blood cells, as beta-thalassemia causes red blood cells to die faster)
- Infections (cause by a malfunctioning spleen)
β-thalassemia Intermedia
The signs and symptoms of β-thalassemia Intermedia usually appear in early childhood or later. Affected people with β-thalassemia Intermedia have mild to moderate anemia, and may also have growth retardation and bone abnormalities. Their symptoms are less severe than beta-thalassemia major and they do not require regular blood transfusions to survive. They may also develop further symptoms (such as an enlarged spleen, gallstones, leg ulcers and thrombosis), due to inefficient production and destruction of red blood cells.
β-thalassemia Minor
People with β-thalassemia Minor only have one defective copy of the
HBB gene and one normal copy. They typically do not show symptoms, but some may have mild anemia.
References:
Origa R (2000, Updated 2015 May 14). Beta-Thalassemia. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews®