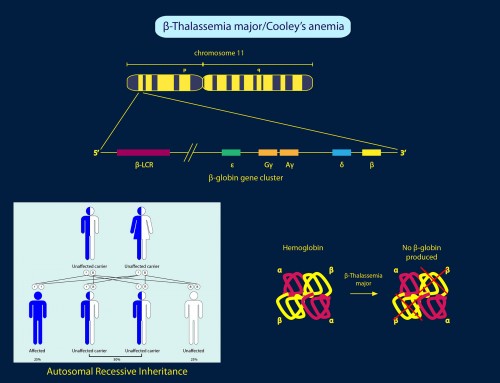
What causes Beta-Thalassemia?
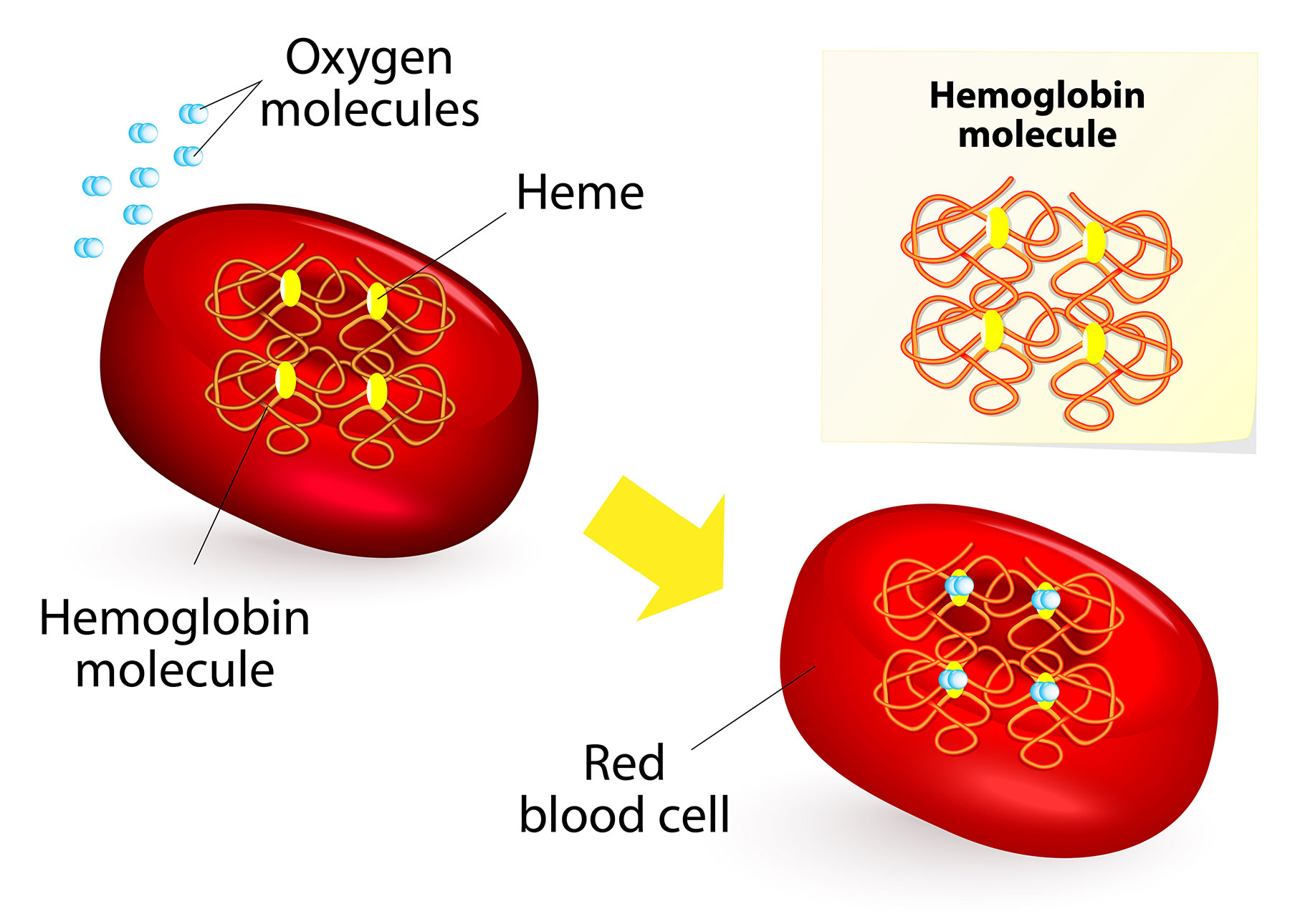
Beta-thalassemia is caused by genetic mutations in the HBB gene which affect the production of β-globin. Insufficient β-globin decreases the amount of functional hemoglobin (HbA), thereby decreasing the amount of red blood cells for maturation. This can lead to anemia and other symptoms in people with beta-thalassemia.
More than 300 mutations in the HBB gene have been identified that cause beta-thalassemia. Most of the mutations involve single nucleotide change, insertions and deletions in or near the HBB gene. HBB gene mutations that eliminate β-globin production result in β0 thalassemia; whereas HBB gene mutations that reduce β-globin production result in β+ thalassemia.
The severity of beta-thalassemia depends on the amount of β-globin produced, which is determined by the type of HBB genetic mutation (β0 or β+) and whether one or two HBB genes are affected. People with only one defective copy of HBB are known as silent carriers of beta-thalassemia. They are usually clinically asymptomatic, as they can make sufficient HbA from the other normal copy of HBB. People who inherit two defective copies of the HBB gene will display symptoms of beta-thalassemia, with varying degrees of severity due to the amount of β-globin produced from the mutant genes.
References:
Origa R (2000, Updated 2015 May 14). Beta-Thalassemia. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2016.
Cao A, Galanello R (2010). Beta-thalassemia. Genetics in Medicine 12, 61–76;
Muncie HL, Campbell JS (2009). Alpha and Beta Thalassemia. Am Fam Physician. 80(4): 339-344.
DNA In the News2017-04-06T20:26:59+00:00